
The USFDA has authorized India to run 703 USFDA-approved pharmaceutical manufacturing facilities, the highest count outside the United States.
The country also oversees 386 EU-GMP compliant plants and 241 WHO-GMP approved plants, according to the 2024 Economic Survey.
USFDA inspection Indian API plants, programmes, and EMA GMP audit Indian pharmaceutical manufacturers assess whether the world’s generic drug supply is safe or not.
The results will determine if the API plant supplier is legally permitted to send the API to you, if the ANDA and the API plant supplier’s product are submitted and supplied to the market.
The FDA FY2024 Report of the State of Pharmaceutical Quality states that it completed 972 drug quality assurance inspections, an increase of 27% from FY2023, and that 62% of those inspections were conducted in other countries.
Indian API Plants Are Prolific, But Inspection Risk Is Concentrated and Consequential
India is the world’s primary API supplier and inspection risks are something no buyer can afford to ignore.
The Scale of India’s API Export Dominance
India’s pharmaceutical industry scale is large. India produces over 500 different active pharmaceutical ingredients (APIs) and accounts for 57% of the APIs in the WHO Prequalified Medicines list (Pharmexcil, 2024). India ranks third in the world in producing pharmaceuticals.
According to the Economic Survey 2024, India’s pharmaceutical exports in the 2024-2025 financial year were $30.47 billion, reflecting a 9.4% year-on-year increase, with exports to the US surpassing $8 billion, also representing a 15% increase.
The Inspection Landscape Why FDA and EMA Scrutiny on India Is Increasing
The USFDA inspection Indian API plants programme and EMA GMP audit Indian pharmaceutical manufacturers schedules are both expanding.
Active inspections of their APIs worldwide, India stands as the largest foreign country supplying APIs to both markets, and gets a large number of their inspections.
According to the Medicine Maker (September 2025), in the fiscal year 2024 of the FDA, India had a huge inspection outcome with 87% of the inspections being in the No Action Indicated and in the Voluntary Action Indicated, while for Europe it was 98%.
The 11% gap in compliance is of concern to the FDA and to the suppliers further along the compliance chain.
Redica Systems has reported that in mid-2024, 41 Indian API and pharmaceutical sites were listed on the FDA’s Import Alert 66-40 list due to a GMP compliance issue.
Of these 41 sites, 21 had no associated Warning Letter confirming that absence of a Warning Letter is not a reliable compliance signal for buyers.
This is the core of the FDA import alert Indian pharma company buyer checklist problem: most buyers do not check proactively.

What Triggers FDA and EMA Inspections of Indian API Plants
Sites with ‘No Application’ status (non-ANDA/NDA) have grown 11% over 5 years, creating a large pool of sites with limited regulatory history that face heightened PAI scrutiny.
| SECTION SUMMARY: India produces 57% of pharmaceuticals APIs that are accredited by the WHO and spends $30.47 billion every year on pharmaceuticals exports. From FY2023 to FY2024, the US FDA increased the number of inspections to Indian pharmaceutical companies by 27%. This increase results in Indian compliance by 11% compared to the level in Europe. 41 Indian companies are listed on the FDA Import Alert 66-40 list, and most of them have never received a Warning Letter. Inspections are conducted based on the assessment of risk, PAI submissions, SIP or other for-cause inspections, and referrals based on the evaluation of the EU EMA. Now we can show the true costs that the buyers have to endure if things go wrong after we checked the scale of India’s dominance in the API market and the inspections. |
The Real Cost of Sourcing from a Non-Compliant Indian API Supplier
Three active consequences, each documented, and all commercially catastrophic to any buyer that does not qualify their API supplier.
An Import Alert Halts Your Entire Supply Chain Instantly
An FDA Import Alert 66-40 means when an Indian API plant’s FDA compliance 2024 2025 failure occurs, the site is placed on the FDA’s website, and there is no-exceptions Detention Without Physical Examination (DWPE) of their purchases at all US ports.
The API already purchased will be frozen at the port, and if there is no qualified alternate, production will cease.
It takes an estimated 6 to 24 months for the FDA to confirm the violation is corrected, resulting in a supply chain shutdown.
A Warning Letter Can Invalidate Your Regulatory Submission
If your ANDA or NDA references an API supplier who later receives an FDA Warning Letter, the FDA may suspend your application.
The FDA Group suggests that such stoppages last between 12 and 36 months. FY2024 saw a rise in Warning Letters globally.
Quality remained 93% compliant globally but the 7% non-compliant segment includes high-consequence cases concentrated in API suppliers for compounding pharmacies, OTC manufacturers, and ophthalmic products.
If the application is for a product with a $50 million annual revenue potential, the 24-month stoppage equals $100 million in a first-to-market opportunity.
The supplier’s non-compliance is the buyer’s financial loss in the market.
The Documentation Failure That No One Catches Until It’s Too Late
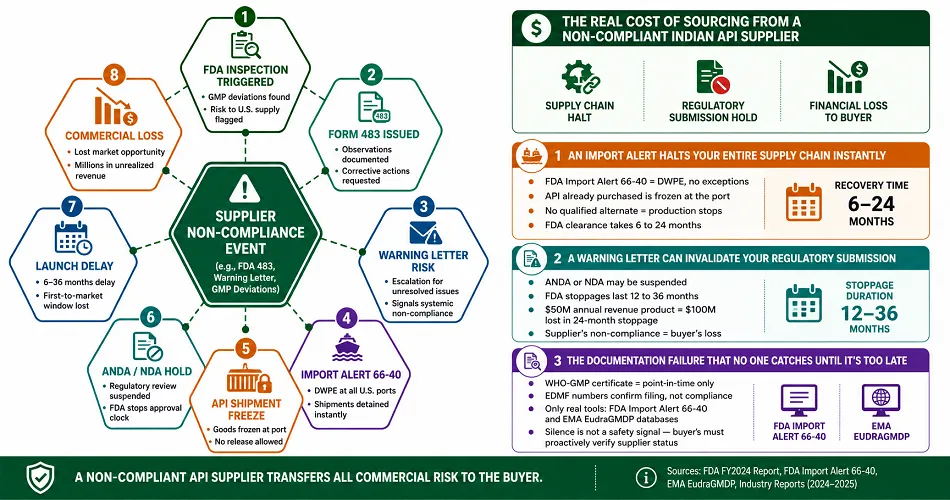
The WHO-GMP certificate API supplier verification shows a supplier who is certified by a WHO-GMP certificate or by the supplier’s EDMF number.
A WHO-GMP certificate only shows compliance then and does not account for time period enforcement by the FDA or EMA that may take place after its issue.
The only tools available to the buyer to assess the time period compliance status of the supplier’s products post issue are the FDA’s Import Alert 66-40 database and EMA’s EudraGMDP database.
| SECTION SUMMARY: FDA Import Alert 66-40 causes detention in all supply chains, with no order values, 6-24 months to sort. A supplier Warning Letter can shut down your ANDA/NDA at a cost of over $100M in first-mover sales. GMP certificates and EDMF numbers provide a picture of compliance, capturing a moment in time. Customers must monitor FDA Import Alert 66-40 and EMA EudraGMDP self-disclosure by a supplier must not be trusted. Having established the costs of non-compliance, let us turn to what genuine compliance entails in a well established Indian API factory. |
What Genuine Compliance Looks Like Inside an Indian API Plant
This section presents a model of how things should be so that buyers may frame the model and the questions needed to assess their supplier.
The core of compliance is only obtainable by viewing it from the inside.
ICH Q7 as the Operational Backbone
ICH Q7 GMP compliance API manufacturing describes the Good Manufacturing Practice (GMP) guidelines for the production of active pharmaceutical ingredients (API).
It has also been incorporated by the FDA in the 21 CFR Part 211, by the EMA in the EU GMP Part II, and by all the member authorities of PIC/S.
A compliant Indian API plant does not merely hold ICH Q7 as a reference document.
It operationalises it, promoting India CDSCO FDA EU GMP harmonisation at every production gate, from the receipt of raw materials to the final release of the API.
The #1 Reason Indian API Plants Fail FDA Inspections
The number one reason for the Indian API manufacturers’ for the failure is the Data integrity pharma API supplier FDA failures are the single most cited reason that has received a FDA warning letter in the last few years. The FDA finds the same issues over and over again.
This includes backdating lab notebooks and other records, deleting OOS records prior to transcribing them, using correction fluid on a batch record rather than doing a proper single line strike through, and using electronic data systems without an audit trail.
What compliant data integrity looks like: Compliant data integrity follows ALCOA+ principles: data must be attributable, legible, contemporaneous, original, and accurate, plus complete, consistent, enduring, and available throughout its lifecycle for regulatory compliance.
The ALCOA+ principles include the requirements of data documentation to be Complete, Consistent, Enduring, and Available.
These are the operational components to Data Integrity in pharmaceutical API supplier data integrity, compliance governed by the FDA.
How Compliant Indian API Plants Prepare for Inspections
How Indian API manufacturers prepare for FDA inspection sets the best facilities apart from the rest.
Leading Indian API exporters maintain perpetual inspection readiness, not compliance sprints before scheduled audits.
| SECTION SUMMARY: All the FDA, EMA, and PIC/S require the application of ICH Q7, the only GMP standard they all rely upon. Indian API warning letters are mainly as a consequence of data integrity issues like OOS deletion, backdating and audit trails omissions. Plants without issues do mock inspections without fail each quarter and do not carry out pre-audit sprints in a desperate attempt to avoid failures. ICH Q7 hinges the presence of a Quality Unit, validated methods, and the APPs. With this comprehensive view regarding internal compliance, we now turn to the Indian API suppliers and can identify in detail the compliance criteria for each of the regulated markets. |
Compliance Requirements Market by Market: What Indian API Suppliers Must Satisfy
Approval in the US market does not mean approval in the EU, UK, or Australia. Each regulated jurisdiction has its own unique system for submissions, certificates, and inspections.
Regulated Market Requirements Comparison
| Market | Key Filing / Certificate | Compliance Body | Key Risk |
|---|---|---|---|
| USA (FDA) | Type II DMF; no active Import Alert 66-40; 21 CFR Part 211 cGMP | CDER / FDA | Import Alert, Warning Letter, PAI failure |
| EU (EMA) | CEP from EDQM preferred; EDMF accepted; GMP cert from PIC/S authority | EMA / Member NCA | EudraGMDP non-compliance listing |
| UK (MHRA) | ASMF + UK MHRA GMP certificate separate from EU post-Brexit | MHRA | EU certificate does not cover UK market |
| Australia (TGA) | TGA overseas GMP clearance not automatic from FDA or EMA certificate | TGA | Clearance takes 3-6 months minimum |
| Japan (PMDA) | JGMP compliance; Japanese language dossier; local MAH mandatory | PMDA | Local agent adds 6-12 months to timeline |
| WHO-PQ | Full WHO API Prequalification certificate for PEPFAR / Global Fund procurement | WHO PQT | Cannot be substituted by FDA or EMA approval |
| Canada (HC) | HC-recognised authority cert | Health Canada | HC may not recognise FDA/EMA inspection if site not in PICS/MRA scope |
What Buyers in Semi-Regulated Markets (ASEAN, LATAM, Africa) Must Check
Argentina’s ANMAT has an API registry. Indian API suppliers without local agent support or local source registration face systematic barriers to LATAM procurement but those with local registration in place through ANVISA, INVIMA, or ANMAT are active in these markets.
| SECTION SUMMARY: Seven fully independent compliance frameworks are the US, EU, UK, Australia, Canada, Japan, and WHO-PQ and none are automatically transferrable. The UK post-Brexit does not accept EU approval and necessitates both an ASMF and an MHRA GMP certificate. Health Canada does not guarantee that the FDA or EMA inspections of establishments that fall outside the PIC/S MRA scope are recognized. Stricter API source requirements are being adopted by semi-regulated markets in the ASEAN, LATAM, and African regions. After establishing a comprehensive understanding of the requirements in each market. We are now able to create a step by step qualification framework that we can then give to procurement teams to implement with immediacy. |
The Buyer’s Framework: How to Qualify an Indian API Supplier for Regulated Market Sourcing
It should come as no surprise that compliant Indian API suppliers do exist.
However, identifying compliant suppliers among non-compliant suppliers demands a systematic and methodical process to help achieve that objective.
The 5 steps defined below aim to address the most frequently encountered buyer-related qualification challenges.
The 5-Step Indian API Supplier Qualification Protocol
Real-time analysis of the supplier’s current regulatory status can be conducted in the FDA’s Import Alert 66-40 and EMA’s EudraGMDP databases. Both databases are public and contain information in real-time.
If a supplier is found to be in any of these databases, they are immediately disqualified.
This is step one of any FDA import alert Indian pharma company buyer checklist.
This analysis must be repeated at least biannually and should aim to capture the supplier’s self-disclosure as a primary tool.
Current GMP certificate (less than 2 years), DMF, EDMF, or CEP number (confirmed current filing status), and a WHO-PQ listing.
Also, it is important to do WHO-GMP certificate API supplier verification.
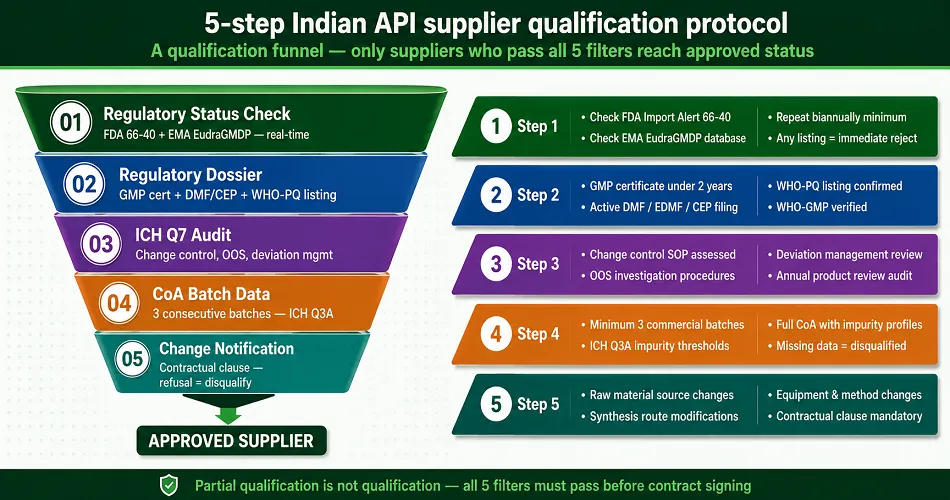
Change control SOP, Out of Specification Investigations (OOS), deviation management, and annual reviews must be assessed against ICH Q7 GMP compliance API manufacturing standards.
ICH Q3A threshold – CoA or impurity data attributed to a specification. An absence of data or an omission of an ICH Q3A threshold.
Any supply agreement must include mandatory notifications for any changes in the raw material source.
Assess data integrity, pharma API supplier, FDA standards, the synthesis route, the equipment, and the testing method. Any supplier who objects to this clause is effectively the biggest hidden risk in API procurement.
Supply Chain Resilience: Building a Dual-Source Indian API Strategy
The HHS ASPE Drug Shortages Analysis January 2025 states that in 2020, manufacturing and quality issues caused 62% of all drug shortages in the US, and by August 2024, only 24% of API manufacturing plants of drugs for the US market were in the US.
A large Indian API manufacturer that has FDA, EU-GMP approval, and a current DMF/CEP, select a manufacturer with verifiable regulatory approvals with API supplier verification compliant with WHO-GMP, and a WHO-PQ listed supplier.
A smaller WHO-GMP-certified Indian supplier or an EU/Israeli/South Korean alternative, who is qualified, but currently on hold and has at least one successful test batch declared.
80/20 in regular processing; a trigger-based shift to 60/40 if the primary supplier is issued a Form 483 with considerate comments.
Commit to a full contract right to audit, with or without the right to audit at a distance, with a 30-day notification clause. Any supplier that refuses to grant audit rights is eliminated from the list of options.
| SECTION SUMMARY: 5-step qualification: real-time status check, dossier assessment, ICH Q7 evaluation, multi-batch CoA, and change-of-contract notification. Policy on change notification clauses is a disqualifying concern, not a negotiable issue. 62% of drug shortages in the US are attributed to subpar manufacturing and quality. A dual-source architecture is imperative. Primary (FDA/EU approved) + secondary (qualified standby) with an 80/20 volume split and trigger-based rebalancing. With the qualification framework firmly in place, the next phase is examining the expected changes in the Indian API compliance landscape from 2025 to 2030. |
India API Compliance Landscape 2025-2030: What Buyers Should Expect
India’s API manufacturing has scope for improvement, despite the country’s amendments.
India’s revised Schedule M amendments mandate WHO-GMP alignment for all Indian pharmaceutical manufacturers by phased timelines through 2025–2026 raising the domestic GMP baseline, though FDA compliance still requires separate FDA inspection and approval.
India’s Structural GMP Improvement Trajectory
India’s Manufacturing Incentives (PLI) program has 261 API manufacturing sites planned as of the June, 2024 Economic Survey, with many constructed for compliance with regulated market export.
The annual US FDA report shows the compliance and success rates for India have steadily improved due to consistent API inspections and scrutiny.
Three scenarios for buyers from 2025 to 2030 regarding the API compliance in India are:
Risk-Adjusted Scenarios for API Buyers Sourcing from India
| Scenario | Conditions | Impact on Buyers | Recommended Strategy |
|---|---|---|---|
| Optimistic (2025-2027) | India and EU converge on MRA GMP; FDA inspection schedules normalize; Schedule M drives industry-wide GMP improvements | Greater availability of Indian API manufacturers; reduced qualification risk; improved cost competitiveness | Maintain qualification standards while expanding sourcing to leverage cost advantages |
| Base Case (2025-2028) | Moderate compliance improvements; FDA inspections continue with limited repeat violations | Stable confidence in supplier qualification; reliance on dual sourcing; ~12–15% of Indian API suppliers remain key | Continue current 5-step qualification protocol with annual re-verification |
| Pessimistic (2026-2028) | US-India trade tensions; stricter FDA compliance; Schedule M delays for smaller manufacturers | Higher qualification burden; potential temporary bans on API suppliers | Strengthen dual sourcing beyond India; maintain 6 months of safety stock; prioritize multi-authority approved suppliers |
| SECTION SUMMARY: Schedule M and PLI Scheme build a comprehensive baseline for Indian API GMP compliance for 2025-2026. Optimistic scenario: India-EU MRA deadline and Schedule M drive price growth. Base case: moderate improvement, dual-source strategy, 12-15% sites remain high-risk, and moderate gap closing. Pessimistic scenario: US-India trade tensions and smaller-manufacturer gaps, six months’ safety stock recommended. Now, let’s answer the most common operational questions procurement and regulatory teams have when qualifying Indian API suppliers. |
FAQ Section
Q1: How do I check if an Indian API supplier has an active FDA Import Alert?
The FDA Import Alert database is publicly accessible at FDA official website and is updated on a rolling basis to check it proactively, not just at onboarding. You can never trust a supplier to inform their client if there is an FDA Import Alert in response to their enforcement.
Q2: What is the difference between a Form 483 and an FDA Warning Letter?
A Form 483 is a way that the FDA communicates after an inspection that there were deficiencies and what those deficiencies were. It is a part of the internal enforcement mechanism of the FDA, and before there is any sort of escalation, the facility can issue a compliance response.
If the FDA is unhappy with that response, it is good to find the deficiencies of a more serious nature; the FDA, at its discretion, may issue a Warning Letter. A Warning Letter may also initiate a halt on applications and importation of products.
Q3: Is a current WHO-GMP certificate sufficient to confirm an Indian API supplier’s compliance for FDA or EMA submissions?
No. A WHO-GMP certificate confirms compliance at the time of inspection only it does not reflect subsequent findings by the FDA or EMA. Only the FDA Import Alert 66-40 database and EMA EudraGMDP provide real-time compliance status. Always verify both databases independently, regardless of what certificates a supplier presents.
Q4: How long does it take for an Indian API supplier to be removed from the FDA Import Alert?
Generally, removal from FDA Import Alert 66-40 takes over 6 months to 2 years or more. The facility must show via a follow-up inspection or documented corrective action, such as monitoring the FDA import alert Indian pharma company buyer checklist. The FDA does not provide a timeline for removal.
Q5: What documents should an Indian API supplier provide before the first shipment to a regulated market buyer?
The documentation includes: a current GMP certificate from the last 24 months (applying WHO-GMP certificate API supplier verification) an active DMF, EDMF, or CEP; a Site Master File; the last three commercial batches along with the complete CoAs including impurity breakdown; a clean record in both the FDA Import Alert database and EMA EudraGMDP; and a signed supply agreement that includes a change notifications clause are helpful in verifying data integrity pharma API supplier FDA standards.
Q6: Does an Indian API supplier need separate qualifications for FDA and EMA market submissions?
Yes, totally. The EMA and FDA have different GMP standards, different filing systems, and different inspection instances. This is the reason why India CDSCO FDA EU GMP harmonisation matters. The US requires a Type II DMF with 21CFRPart211 compliance, while the EU requires a CEP or EDMF and an EU-approved GMP certificate. These are fully independent certifications.
Q7: What is ICH Q7, and why should API buyers-not just manufacturers – care about it?
ICH Q7 GMP compliance API manufacturing formally the ICH Q7 guideline is the internationally agreed GMP standard for API manufacturing. It covers raw material sourcing, change control, data integrity, pharma API supplier, FDA requirements, validation, and Quality Unit independence.
Every USFDA inspection of Indian API plants assessment, and every EMA GMP audit of the Indian pharmaceutical manufacturers programme is conducted with ICH Q7 as the primary reference standard. Buyers who understand ICH Q7 can ask the right qualification questions distinguishing a genuinely robust quality system from one that is merely certificated.
Conclusion
India has 703 USFDA-approved manufacturing units and has a 57% contribution to the WHO Prequalified API supply.
It has USD 30.47 billion in annual pharmaceutical exports, according to the Economic Survey 2024.
Because of these aspects, India is the primary supplier of global generic medicine.
The FDA FY2024 Annual Report shows India’s 87% favorable outcome rate but still trails Europe’s 98%, and 41 Indian sites sit on FDA Import Alert 66-40.
The compliance landscape is active, evolving, and progressing.
Buyers who succeed are those who verify supplier status in real-time, create contractual change notification obligations, and construct dual-source architecture capable of surviving supply disruption.
Indian API manufacturers can provide the highest level of supply security. That is, for buyers who understand how to find, qualify, and protect Indian supply.
However, the Progress on India CDSCO, FDA EU GMP harmonisation is on the move.
Would you like to evaluate your Indian API supplier against the five-step qualification protocol? The FDA Import Alert 66-40 alone could answer your question.
It could take you under five minutes, and could end up being the most important five minutes in your next regulatory submission.